












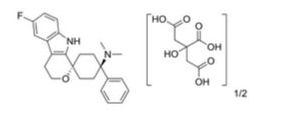
Cebranopadol hemicitrate, GRT-6005
Phase III
Grünenthal GmbH innovator
SYNTHESIS COMING WATCH OUT……….
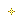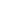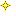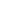
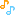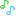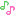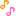

A mu-opioid agonist for treatment of neuropathic pain and pain due to osteoarthritis.
![]()
CAS No.863513-92-2(Cebranopadol Hemicitrate)
CAS 863513-91-1(FREE FORM)
Spiro[cyclohexane-1,1′(3’H)-pyrano[3,4-b]indol]-4-amine, 6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-, trans
![]()
Cebranopadol (GRT-6005) is a novel opioid analgesic of the benzenoid class which is currently under development internationally by Grünenthal, a German pharmaceutical company, and its partner Depomed, a pharmaceutical company in the United States, for the treatment of a variety of different acute and chronic pain states.[1][2][3] As of November 2014, it is in phase III clinical trials. Cebranopadol is unique in its mechanism of action as an opioid, binding to and activating all four of the opioid receptors; it acts as afull agonist of the nociceptin receptor (Ki = 0.9 nM; EC50 = 13.0; IA = 89%), μ-opioid receptor (Ki = 0.7 nM; EC50 = 1.2; IA = 104%), and δ-opioid receptor (Ki = 18 nM; EC50 = 110; IA = 105%), and as a partial agonist of the κ-opioid receptor (Ki = 2.6 nM; EC50 = 17; IA = 67%).[1] The ED50 values of 0.5-5.6 µg/kg when introduced IV & 25.1 µg/kg after oral administration.[4]
![]()
Cebranopadol shows highly potent and effective antinociceptive and antihypertensive effects in a variety of different animal modelsof pain.[1] Notably, it has also been found to be more potent in models of chronic neuropathic pain than acute nociceptive paincompared to selective μ-opioid receptor agonists.[1] Relative to morphine, tolerance to the analgesic effects of cebranopadol has been found to be delayed (26 days versus 11 days for complete tolerance).[1] In addition, unlike morphine, cebranopadol has not been found to affect motor coordination or reduce respiration in animals at doses in or over the dosage range for analgesia.[1] As such, it may have improved and prolonged efficaciousness and greater tolerability in comparison to currently available opioid analgesics.[1]

As an agonist of the κ-opioid receptor, cebranopadol may have the capacity to produce psychotomimetic effects and other adverse reactions at sufficiently high doses, a property which could potentially limit its practical clinical dosage range.[5]
Cebranopadol (trans-6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3′H)-pyrano[3,4-b]indol]-4-amine) is a novel analgesic nociceptin/orphanin FQ peptide (NOP) and opioid receptor agonist [Ki (nM)/EC50(nM)/relative efficacy (%): human NOP receptor 0.9/13.0/89; human mu-opioid peptide (MOP) receptor 0.7/1.2/104; human kappa-opioid peptide receptor 2.6/17/67; human delta-opioid peptide receptor 18/110/105]. Cebranopadol exhibits highly potent and efficacious antinociceptive and antihypersensitive effects in several rat models of acute and chronic pain (tail-flick, rheumatoid arthritis, bone cancer, spinal nerve ligation, diabetic neuropathy) with ED50 values of 0.5−5.6 µg/kg after intravenous and 25.1 µg/kg after oral administration. In comparison with selective MOP receptor agonists, cebranopadol was more potent in models of chronic neuropathic than acute nociceptive pain. Cebranopadol’s duration of action is long (up to 7 hours after intravenous 12 µg/kg; >9 hours after oral 55 µg/kg in the rat tail-flick test). The antihypersensitive activity of cebranopadol in the spinal nerve ligation model was partially reversed by pretreatment with the selective NOP receptor antagonist J-113397[1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one] or the opioid receptor antagonist naloxone, indicating that both NOP and opioid receptor agonism are involved in this activity. Development of analgesic tolerance in the chronic constriction injury model was clearly delayed compared with that from an equianalgesic dose of morphine (complete tolerance on day 26 versus day 11, respectively). Unlike morphine, cebranopadol did not disrupt motor coordination and respiration at doses within and exceeding the analgesic dose range. Cebranopadol, by its combination of agonism at NOP and opioid receptors, affords highly potent and efficacious analgesia in various pain models with a favorable side effect profile.
Almost 20 years ago, a new member of the opioid receptor family and its endogenous agonist were described (Meunier et al., 1995; Reinscheid et al., 1995). Because of its partial homology to the opioid receptors [mu-opioid peptide (MOP) receptor, delta-opioid peptide (DOP) receptor, kappa-opioid peptide (KOP) receptor] and its insensitivity to the prototypical opioid agonist and antagonist ligands morphine and naloxone, this receptor was initially termed opioid receptor-like receptor, ORL1. Subsequently, it was renamed the nociceptin/orphanin FQ peptide (NOP) receptor after its endogenous ligand nociceptin, and it is now considered to be a non-opioid member of the opioid receptor family (Cox et al., 2009). At a cellular level, the actions of the NOP receptor are broadly similar to those of the opioid receptors (Chiou et al., 2007; Lambert, 2008). Although NOP receptors are clearly expressed at all levels of the pain pathways, it is thought that NOP and MOP receptors are not colocalized in the same neurons and may, thus, have independent actions in at least partly distinct neuronal networks (Monteillet-Agius et al., 1998).
The role of the NOP receptor in pain and analgesia has remained unclear for some time owing to inconsistent findings in early reports using nociceptin to activate the receptor. Being a peptide, nociceptin was administered locally into the central nervous system (CNS) where it produced both pronociceptive and antinociceptive effects when administered supraspinally (Meunier et al., 1995; Calo and Guerrini, 2013). Remarkably, when administered into the spinal cord of rodents and nonhuman primates, nociceptin consistently produced antinociceptive effects (Ko et al., 2009; Sukhtankar and Ko, 2013). Subsequent studies of systemic administration of nonpeptide NOP receptor agonists revealed that such compounds were effective analgesics in animal pain models. Although evidence for antinociceptive and antihyperalgesic effects in rodents is limited and inconsistent (Jenck et al., 2000; Reiss et al., 2008), Ko et al. (2009) demonstrated impressive antinociceptive and antiallodynic potency and efficacy using the NOP receptor agonist Ro64-6198 in Rhesus monkeys. Potency and efficacy were comparable with those of alfentanil but with a complete absence of alfentanil-associated side effects such as itching/scratching and respiratory depression and no evidence of reinforcing effects (Ko et al., 2009; Podlesnik et al., 2011).
Currently, strong MOP receptor agonists are the most effective drugs for the treatment of moderate to severe acute and chronic pain. However, although these drugs provide potent analgesia, they also carry the risk of severe side effects such as respiratory depression, nausea, vomiting, and constipation, and their use may lead to physical dependence and tolerance (Zöllner and Stein, 2007). In addition, opioids are considered to have limited efficacy in treating chronic nociceptive and neuopathic pain owing to a reduction in the already low therapeutic index (Rosenblum et al., 2008; Labianca et al., 2012). For these reasons, there is an unmet medical need for potent and well-tolerated analgesics for the treatment of moderate to severe chronic nociceptive and neuropathic pain.
As NOP and opioid receptor agonists modulate pain and nociception via distinct yet related targets, combining both mechanisms may constitute an interesting and novel approach for the development of innovative analgesics. Notably, a supra-additive interaction between intrathecal morphine and intrathecal nociceptin has been described in rodents (Courteix et al., 2004), as well as an enhancement of the antinociceptive effect of systemic morphine by systemic administration of Ro64-6198 (Reiss et al., 2008). Furthermore, a synergistic effect of concurrent NOP and MOP receptor activation without significant side effects has been demonstrated in nonhuman primates after systemic administration (Cremeans et al., 2012). At the same time, activation of NOP receptors has been proposed to counteract supraspinal opioid activity; in animal studies, NOP receptor agonists do not generate typical opioid-like side effects and may even ameliorate opioid-related side effects when administered concurrently with an opioid agonist (Ko et al., 2009; Rutten et al., 2010; Toll, 2013). Thus, a combination of NOP and opioid receptor activation may be particularly suited to provide potent analgesia with reduced opioid-like side effects.
To explore the potential benefits of NOP and opioid receptor coactivation, novel compounds acting as agonists on both NOP and opioid receptors have been designed (Molinari et al., 2013; Zaveri et al., 2013). This article describes the preclinical pharmacology of cebranopadol, a potent NOP and opioid receptor agonist derived from a novel chemical series of spiro[cyclohexane-dihydropyrano[3,4-b]indol]-amines (S. Schunk, K. Linz, C. Hinze, S. Frormann, S. Oberbörsch, B. Sundermann, S. Zemolka, W. Englberger, T. Germann, T. Christoph, B.Y. Kögel, W. Schröder, S. Harlfinger, D. Saunders, A. Kless, H. Schick, and H. Sonnenschein, submitted manuscript) that was developed by Grünenthal (Aachen, Germany) and is currently in clinical development for the treatment of severe chronic pain……..http://jpet.aspetjournals.org/content/349/3/535.full
WO 2013170968
WO 2013170966
WO 2013170971
WO 2013170972
WO 2013170970
WO 2013170969
WO 2013170967
WO 2004043967
US 20130150590


PAPER
ACS Medicinal Chemistry Letters (2014), 5(8), 857-862.
Discovery of a Potent Analgesic NOP and Opioid Receptor Agonist: Cebranopadol
Abstract

In a previous communication, our efforts leading from 1 to the identification of spiro[cyclohexane-dihydropyrano[3,4-b]indole]-amine 2a as analgesic NOP and opioid receptor agonist were disclosed and their favorable in vitro and in vivo pharmacological properties revealed. We herein report our efforts to further optimize lead 2a, toward trans-6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3′H)-pyrano[3,4-b]indol]-4-amine (cebranopadol, 3a), which is currently in clinical development for the treatment of severe chronic nociceptive and neuropathic pain.
http://pubs.acs.org/doi/abs/10.1021/ml500117c?source=chemport&journalCode=amclct

MP 258-282 DEG CENT


October the family Grünenthal GmbH celebrated its longtime employee in Aachen-Eilendorf. Proud 680 years of service …
References
- Linz K, Christoph T, Tzschentke TM; et al. (June 2014). “Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist”. J. Pharmacol. Exp. Ther. 349 (3): 535–48. doi:10.1124/jpet.114.213694.PMID 24713140.
- Schunk S, Linz K, Hinze C; et al. (August 2014). “Discovery of a Potent Analgesic NOP and Opioid Receptor Agonist: Cebranopadol”. ACS Med Chem Lett 5 (8): 857–62.doi:10.1021/ml500117c. PMID 25147603.
- Lambert DG, Bird MF, Rowbotham DJ (September 2014). “Cebranopadol: a first in-class example of a nociceptin/orphanin FQ receptor and opioid receptor agonist”. Br J Anaesth114: 364–6. doi:10.1093/bja/aeu332. PMID 25248647.
- Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. Journal of Pharmacol Exp Ther. 2014 Jun;349(3):535-48. doi: 10.1124/jpet.114.213694
- Pfeiffer A, Brantl V, Herz A, Emrich HM (August 1986). “Psychotomimesis mediated by kappa opiate receptors”. Science 233 (4765): 774–6. doi:10.1126/science.3016896.PMID 3016896.
- Expert Opinion on Investigational Drugs (2015), 24(6), 837-844
- Journal of Pharmacology and Experimental Therapeutics (2014), 349(3), 535-548,
-
External links
|
|
| Systematic (IUPAC) name | |
|---|---|
|
(1r,4r)-6’-fluoro-N,N-dimethyl-4-phenyl-4’,9’-dihydro-3’H-spiro[cyclohexane-1,1’-pyrano[3,4-b]indol]-4-amine
|
|
| Pharmacokinetic data | |
| Biological half-life | ~4.5 hours |
| Identifiers | |
| CAS Number | 863513-91-1 |
| ATC code | None |
| PubChem | CID 11848225 |
| ChemSpider | 29398942 |
| Chemical data | |
| Formula | C24H27FN2O |
| Molar mass | 378.482 g/mol |
////Cebranopadol hemicitrate, GRT-6005, Cebranopadol, セブラノパドール
CN([C@]1(CC[C@]2(OCCc3c2[nH]c4c3cc(cc4)F)CC1)c5ccccc5)C
