





Iloprost (ciloprost)
| MF | C22H32O4 |
|---|---|
| Formula Wgt | 360.5 |
6,9ALPHA-METHYLENE-11ALPHA,15S-DIHYDROXY-16-METHYL-PROSTA-5E,13E-DIEN-18-YN-1-OIC ACID
6,9α-


Iloprost Molecule
ILOPROST (Ventavis®) is used to treat a serious heart and lung disorder called pulmonary arterial hypertension. While iloprost inhalation solution will not cure this disorder, it is designed to improve symptoms and the quality of life. Generic iloprost inhalation solution is not yet available.
Iloprost is a second generation structural analog of prostacyclin (PGI) with about ten-
73873-87-7 CAS NO
78919-13-8 PHENACYL ESTER
Launched – 1992 bayer
Ilomedin®, Ventavis™
 iloprost
iloprost
An eicosanoid, derived from the cyclooxygenase pathway of arachidonic acid metabolism. It is a stable and synthetic analog of EPOPROSTENOL, but with a longer half-life than the parent compound. Its actions are similar to prostacyclin. Iloprost produces vasodilation and inhibits platelet aggregation.
BAY-q-6256 E-1030 SH-401 ZK-36374
- BAY Q6256
- Ciloprost
- Iloprost
- Iloprostum
- Iloprostum [Latin]
- UNII-AHG2128QW6
- UNII-JED5K35YGL
- Ventavis
- ZK 00036374
- ZK 36374
Endoprost Ilomedin Ilomédine Ventavis Iloprost is a synthetic prostacyclin analog discovered and developed by Schering AG and Berlex which has been available for more than ten years as therapy for peripheral arterial occlusive disease (PAOD), including Raynaud’s phenomenon and Buerger’s disease.
Iloprost improves blood flow, relieves the pain associated with circulatory disturbances and improves the healing of ulcers, which can develop as a result of poor arterial blood flow. Iloprost also produces vasodilatation of the pulmonary arterial bed, with subsequent significant improvement in pulmonary artery pressure, pulmonary vascular resistance and cardiac output, as well as mixed venous oxygen saturation. In 2003, Schering AG received approval in the E.U. for an inhaled formulation of iloprost (Ventavis[R]) for the treatment of primary pulmonary hypertension and the following year, the product was launched in Germany and the U.K.
Introduction on the U.S. market took place in March 2005 by CoTherix for the same indication in patients with NYHA Class III or IV symptoms. Iloprost is also available for the treatment of pulmonary hypertension and peripheral vascular disease. CoTherix had been developing a dry powder for potential use in the treatment of pulmonary hypertension; however, no recent development has been reported for this research. In Japan, phase III clinical trials are ongoing for the treatment of pulmonary arterial hypertension.  In 2003, CoTherix licensed exclusive rights from Schering AG to market iloprost in the U.S. for primary pulmonary hypertension while Schering AG retained rights to the product outside the U.S. In April 2005, CoTherix established a collaborative research and development agreement with Quadrant to develop an extended-release formulation of iloprost inhalation solution. Iloprost was designated as an orphan medicinal product for the treatment of pulmonary hypertension in December 2000 by the EMEA and will fall under orphan drug protection until 2013.
In 2003, CoTherix licensed exclusive rights from Schering AG to market iloprost in the U.S. for primary pulmonary hypertension while Schering AG retained rights to the product outside the U.S. In April 2005, CoTherix established a collaborative research and development agreement with Quadrant to develop an extended-release formulation of iloprost inhalation solution. Iloprost was designated as an orphan medicinal product for the treatment of pulmonary hypertension in December 2000 by the EMEA and will fall under orphan drug protection until 2013.
The FDA has assigned to iloprost several orphan drug designations. In 1989, iloprost solution for infusion was granted orphan drug designation for the treatment of Raynaud’s phenomenon secondary to systemic sclerosis followed by another orphan drug designation in 1990 for iloprost solution for injection for the treatment of heparin-associated thrombocytopenia. In 2004, an additional orphan drug designation for iloprost inhalation solution for the treatment of pulmonary arterial hypertension was assigned.
The status has also been assigned in the E.U. for this indication. In 2012, orphan drug designation was assigned in the U.S. for the treatment of purpura fulminans in combination with eptifibatide and for the treatment of pulmonary arterial hypertension. In 2007, Cotherix was acquired by Actelion.
 ILOPROST
ILOPROST
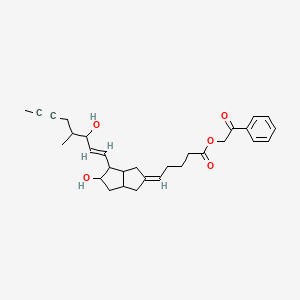
Ventavis (TN), Iloprost phenacyl ester, Iloprost-PE, Iloprost (INN), CHEMBL138694, CHEMBL236025, AC1O6009, DAP000273, CID5311181
2-oxo-2-phenylethyl 5-[(2Z)-5-hydroxy-4-[(1E)-3-hydroxy-4-methyloct-1-en-6-yn-1-yl]-octahydropentalen-2-ylidene]pentanoate
IMPORTANT PUBLICATIONS
Ciloprost Drugs Fut 1981, 6(11): 676
A carbohydrate approach for the formal total synthesis of the prostacyclin analogue (16S)-iloprost Tetrahedron Asymmetry 2012, 23(5): 388
Angewandte Chemie, 1981 , vol. 93, 12 pg. 1080 – 1081
Tetrahedron Letters, 1992 , vol. 33, 52 pg. 8055 – 8056
Helvetica Chimica Acta, 1986 , vol. 69, 7 pg. 1718 – 1727
Journal of Medicinal Chemistry, 1986 , vol. 29, 3 pg. 313 – 315
US5286494 A1
| US 2013253049 |
uS 2013184295
WO 1992014438
WO 1993007876
WO 1993015739
WO 1994008584
WO 2013040068
WO 2012174407
WO 2011047048
| EP0011591A1 * | Oct 18, 1979 | May 28, 1980 | Schering Aktiengesellschaft | Prostane derivatives, their production and pharmaceutical compositions containing them |
| EP0084856A1 * | Jan 19, 1983 | Aug 3, 1983 | Toray Industries, Inc. | 5,6,7-Trinor-4, 8-inter-m-phenylene prostaglandin I2 derivatives |
| EP0099538A1 * | Jul 11, 1983 | Feb 1, 1984 | Schering Aktiengesellschaft | Carbacyclines, process for their preparation and their use as medicines |
……………………………………
-
5,6,7-trinor-4,8-inter-m-phenylene prostaglandin 12derivatives.
-
Prostaglandin I2, hereinafter referred to as PGI2, of
![Figure imgb0001]()
was first found by J.R. Vane et.al. in 1976 and is biosynthe- sized from arachidonic acid via endoperoxide(PGH2 or PGG2) in the vascular wall. PGI2 is well known to show potent activity to inhibit platelet aggregation and to dilate peripheral blood vessels(C & EN, Dec. 20, 1976, page 17 and S. Moncade et al., Nature, 263,633(1976)).
-
[0003]Because of the unstable exo-enolether structure thereof, PGI2 is extremely unstable even in a neutral aqueous solution and is readily converted to 6-oxo-PGF1α which is almost physiologically inactive. Such instability of PGI2 is a big obstacle to its use as a drug. Furthermore, PGI2 is unstable in vivo as well and shows only short duration of action.
-
The compounds of the present invention are novel PGI2 derivatives in which the exo-enolether moiety characteristic of PGI2 is transformed into “inter-m-phenylene” moiety. In this sense the compounds may be regarded as analogs of PGI2.
-
The compounds of the present invention feature much improved stability in vitro as well as in vivo in comparison with PGI2. The compounds are highly stable even in an aqueous solution and show long duration of action in vivo. Further, the compounds have advantages over PGI2 for pharmaceutical application because they exhibit more selective physiological actions than PGI2, which has multifarious, inseperable biological activities.
-
Some prostaglandin I2 derivatives which have 5,6,7-tri- nor-4,8-inter-m-phenylene structure have already been described in publication by some of the present authors. (Kiyotaka Ohno, Hisao Nishiyama and Shintaro Nishio, U.S.P. 4,301,164 (1981)). But, the compounds of the present invention, which feature the presence of alkynyl side chain, have more potent physiological activities as well as decreased side effects than the already disclosed compounds analogous to those of the present invention.
-
It is an object of this invention to provide novel prostaglandin I2derivatives which are stable and possess platelet aggregation-inhibiting, hypotensive, anti-ulcer and other activities.

-
![Figure imgb0004]()
is named as 16-methyl-18,19-tetradehydro-5,6,7-trinor-4,8-inter-m-phenylene PGI2.
-
Alternatively, the compound of the formula (II) may be named as a derivative of butyric acid by the more formal nomenclature. In such a case, the condensed ring moiety is named after the basical structure of 1H-cyclopenta[b]benzofuran of the following formula:
![Figure imgb0005]()
The term “synthetic prostacyclins” as used herein can refer to any prostacyclin that can be prepared via synthetic organic chemistry, including those prostacyclins that are also naturally occurring, such as Prostacyclin (PGI2):
![Figure imgf000025_0001]()
which is also known as Epopreostenol.
Thus, examples of synthetic prostacyclins include, but are not limited to: Prosta
![Figure imgf000025_0002]()
lloprost, which has the structure:
![Figure imgf000025_0003]()
Trepro inil (also known as Rumodolin), which has the structure:
![Figure imgf000025_0004]()
Beraprost, which has the structure:
![Figure imgf000026_0001]()
as well as the esters, stereoisomers, and salts thereof, or other analogues or derivatives of the recited synthetic prostacyclins, such as compounds comprising other aliphatic linker groups linking the carboxylic acid group to the cyclic components of the synthetic prostacyclins, compounds containing additional alkene and/or alkyne bonds, and/or compounds containing additional substituents on the cyclic components of the synthetic prostacyclins.
![Figure imgf000031_0001]()
iloprost, in contrast to PGI.sub.2 a stable prostacyclin derivative, has been known since 1980 by European patent application EP 11591, no other prostacyclin derivative has previously been tested in this indication. It is therefore reasonable to assume that a spontaneous healing is involved in the published case.
It has now been found, surprisingly, that iloprost is effective in the case of cerebral malaria.
For salt formation of iloprost, inorganic and organic bases are suitable, as they are known to one skilled in the art for the formation of physiologically compatible salts. For example, there can be mentioned: alkali hydroxides, such as sodium and potassium hydroxide, alkaline-earth hydroxides, such as calcium hydroxide, ammonia, amines, such as ethanolamine, diethanolamine, triethanolamine, N-methylglucamine, morpholine, tris-(hydroxymethyl)-methylamine, etc.
The β-cyclodextrin clathrate formation takes place according to EP 259468.
The production of iloprost is described in detail in EP 11591.
-
Nileprost iloprost, and a process for preparing these compositions.
-
From EP 11 591 already carbacyclin derivatives of the cytoprotective effect on the gastric and intestinal mucosa, and the myocardial cytoprotection using carbacyclin derivatives is known.
-
It has now been found that iloprost (I) and Nileprost (II)
![Figure imgb0001]()
and their salts with physiologically acceptable bases and cytoprotective effect in the kidney.
-
Forming salts of iloprost and Nileprost inorganic and organic bases are suitable, as are known to those skilled in the formation of physiologically compatible salts known. Examples which may be mentioned are: alkali metal hydroxides, such as sodium and potassium hydroxide, alkaline earth metal hydroxides such as calcium hydroxide, ammonia, amines, such as ethanolamine, diethanolamine, triethanolamine, N-methylglucamine, morpholine, tris (hydroxymethyl) methylamine, etc.
-
The production of iloprost and is described in detail in EP Nileprost 2234 and EP 11591.
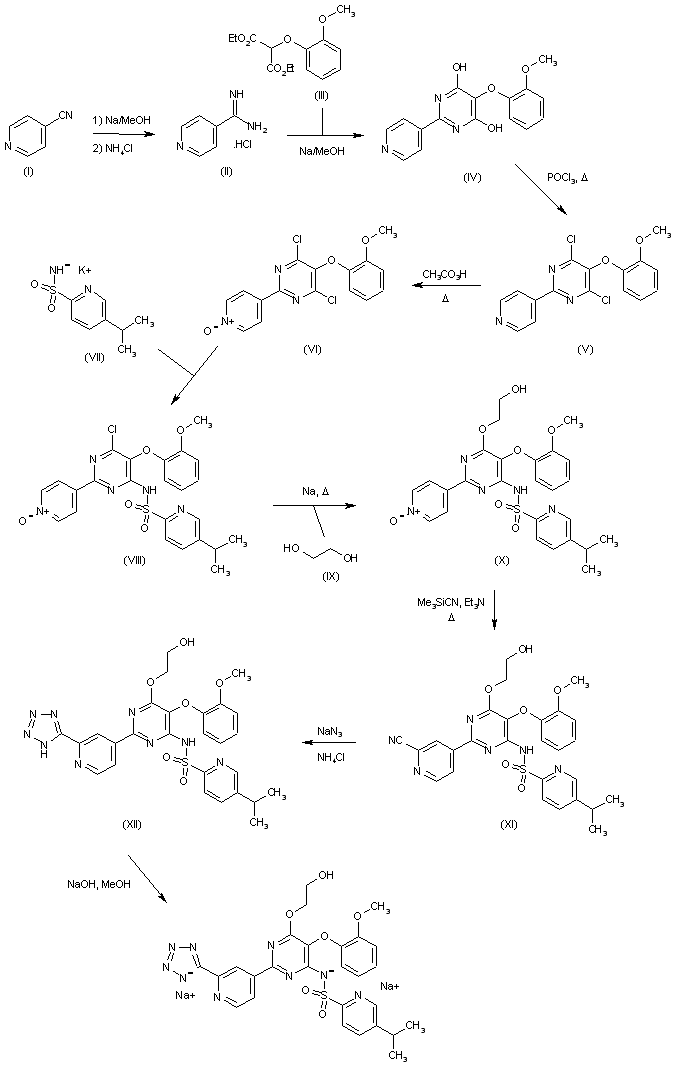
………………..J. Med. Chem., 1986, 29 (3), pp 313–315DOI: 10.1021/jm00153a001 -









see paper

-
Used as the starting material for the method described above ketone of general formula II can be prepared by reacting an alcohol of the formula IV
![Figure imgb0006]()
(EJCorey et al., J. Amer. Chem. 93, 1490 (1971)) transformed with dihydropyran in the presence of catalytic amounts of p-toluenesulfonic acid in the tetrahydropyranyl ether V.
![Figure imgb0007]()
-
[0026]Lactone V with Diisobatylauminiumnydrid reduced at -70 ° C to the lactol VI, which is converted by Wittiereaktion Triphenylphosphoniummethylen with the olefin VII. After conversion to the tosylate with p-toluenesulfonyl chloride in the presence of pyridine is obtained by reaction with potassium nitrite in the dimethylsulfoxide 9SS-configured alcohol IX, which is converted with p-toluenesulfonyl chloride in the presence of pyridine in the tosylate X. Reaction with Malonsäurediäthylester in presence of potassium tert-butoxide gives the diester XI, which is converted by decarbalkoxylation with sodium cyanide in dimethyl sulfoxide in the ester XII.
![Figure imgb0008]()
-
[0027]Oxidative cleavage of the double bond in the compound XII with Hatrium p j o dat it out in the presence of catalytic amounts of osmium tetroxide to give the aldehyde XIII, which is oxidized with Jones reagent to the acid XIV which is then esterified with diazomethane to the compound XV. By Dieckmann condensation of XV with potassium tert-butoxide in tetrahydrofuran is obtained a mixture of isomers of the ketocarboxylic acid ester XVI and XVII, which by means of a decarbalkoxylation with 1,4-diazabicyclo [2,2,2] octane in xylene converted into ketone XVIII as the only reaction product is.
![Figure imgb0009]()
-
[0028]The removal of the Tetrahydropyranylätherschutzgruppe delivers the alcohol XIX, which is esterified with benzoyl chloride in the presence of pyridine to give the ester XX.
![Figure imgb0010]()
-
[0029]Benzyläthers hydrogenolytic cleavage of a catalytic amount of acid gives the alcohol XXI, which is according to ketalization compound XXII oxidized with Collins reagent to aldehyde XXIII.
-
[0030]This aldehyde XXIV with a phosphonate of the general formula
![Figure imgb0011]()
wherein D, E and R 2 have the meanings given above is reacted in a Olefinicrungsreaktion to a ketone of the formula XXV.
![Figure imgb0012]()
-
[0031]After reduction of the 15-keto group with zinc borohydride or sodium borohydride or reaction with alkylmagnesium bromide or alkyllithium and. Epimerentrennung obtain the 15α-alcohols XXVI (PG numbering).
![Figure imgb0013]()
-
[0032]After hydrolysis of the ester group, for example with potassium carbonate in methanol and ketal cleavage with aqueous acetic acid yields the ketone of the formula XXVII,
![Figure imgb0014]()










THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family

 cicaprost
cicaprost cicaprost
cicaprost





 MIDOSTAURIN
MIDOSTAURIN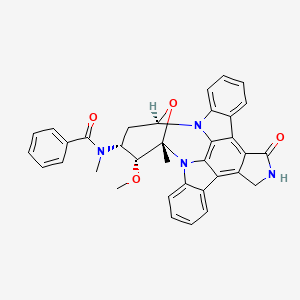 MIDOSTAURIN
MIDOSTAURIN





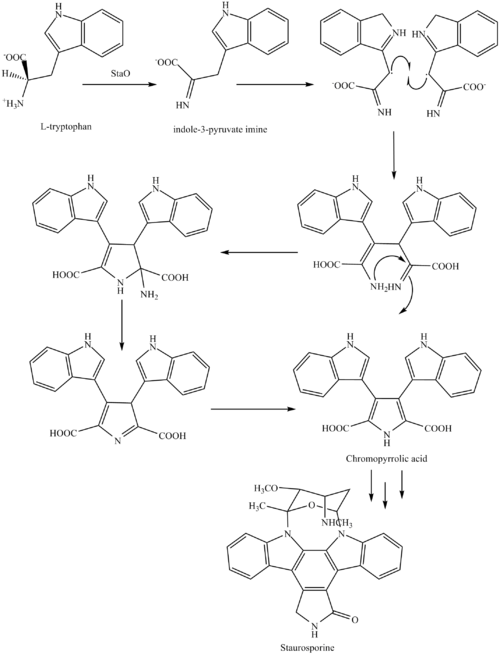


 rapamycin
rapamycin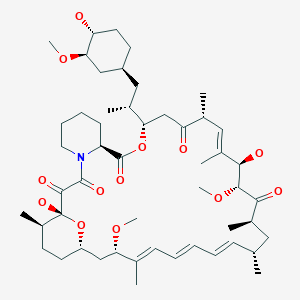 SIROLIMUS
SIROLIMUS











 SIROLIMUS
SIROLIMUS








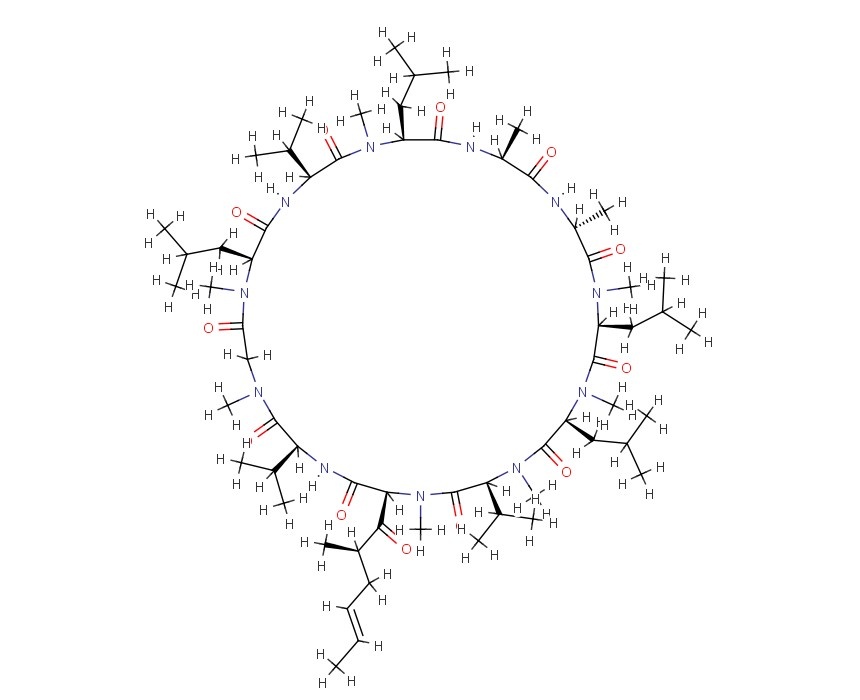


















 CLAZOSENTAN
CLAZOSENTAN CLAZOSENTAN DI-NA SALT is discontinued
CLAZOSENTAN DI-NA SALT is discontinued
















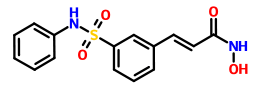
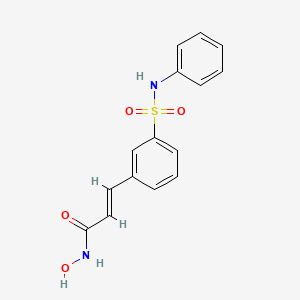 BELINOSTAT
BELINOSTAT









































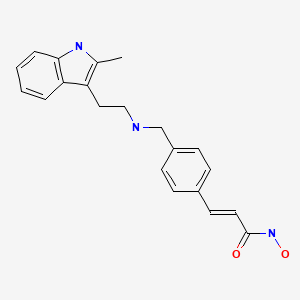 panobinostat
panobinostat













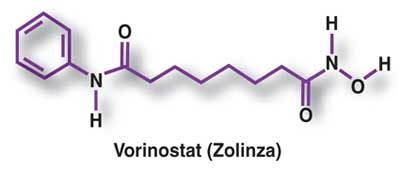
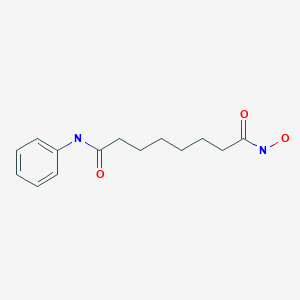 VORINOSTAT
VORINOSTAT



















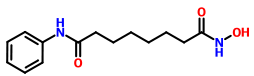

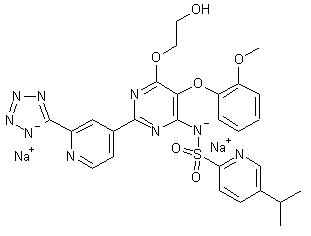
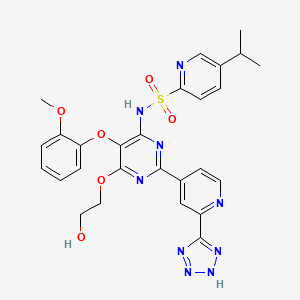 TEZOSENTAN
TEZOSENTAN
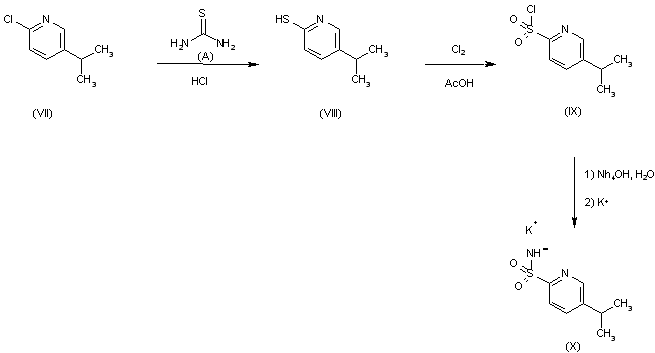
























 DOXOFYLLINE
DOXOFYLLINE





















 DASANTAFIL
DASANTAFIL
















