![Delamanid.svg]()
Delamanid
Otsuka Pharmaceutical Co has been given the green light to sell its tuberculosis drug Deltyba in Europe.
Read more at: http://www.pharmatimes.com/Article/14-04-30/Otsuka_multi-drug_resistant_TB_drug_approved_in_Europe.aspx#ixzz30Sz3wf2k
Follow us: @PharmaTimes on Twitter
http://newdrugapprovals.org/2014/03/26/delamanid-an-experimental-drug-for-the-treatment-of-multi-drug-resistant-tuberculosis/
![Delamanid.svg]()
Delamanid
http://www.ama-assn.org/resources/doc/usan/delamanid.pdf
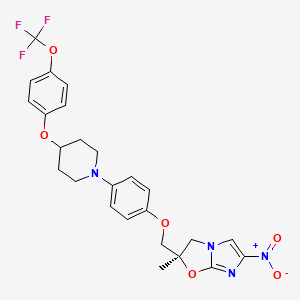
(2R)-2-Methyl-6-nitro-2-[(4-{4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl}phenoxy)methyl]-2,3-dihydroimidazo[2,1-b][1,3]oxazole
2(R)-Methyl-6-nitro-2-[4-[4-[4-(trifluoromethoxy)phenoxy]piperidin-1-yl]phenoxymethyl]-2,3-dihydroimidazo[2,1-b]oxazole
(R) -2-methyl-6-nitro-2- { 4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl } -2 , 3- dihydroimidazo [2 , 1-b] oxazole
Imidazo[2,1-b]oxazole, 2,3-dihydro-2-methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl]phenoxy]methyl]-, (2R)-
(R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole
681492-22-8 cas no
Delamanid, 681492-22-8, Delamanid (JAN/USAN), Delamanid [USAN:INN],UNII-8OOT6M1PC7,
- OPC 67683
- OPC-67683
- UNII-8OOT6M1PC7
Molecular Formula: C25H25F3N4O6
Molecular Weight: 534.48441
CLINICAL TRIALS
Trial Name: A Placebo-Controlled, Phase 2 Trial to Evaluate OPC 67683 in Patients With Pulmonary Sputum Culture-Positive, Multidrug-Resistant Tuberculosis (TB)
Primary Sponsor: Otsuka Pharmaceutical Development & Commercialization, Inc.
Trial ID / Reg # / URL: http://clinicaltrials.gov/ct2/show/NCT00685360
Delamanid (USAN, codenamed OPC-67683) is an experimental drug for the treatment of multi-drug-resistant tuberculosis. It works by blocking the synthesis of mycolic acids in Mycobacterium tuberculosis, the organism which causes tuberculosis, thus destabilising its cell wall.[1][2][3]
![]()
In phase II clinical trials, the drug was used in combination with standard treatments, such as four or five of the drugs ethambutol, isoniazid,pyrazinamide, rifampicin, aminoglycoside antibiotics, and quinolones. Healing rates (measured as sputum culture conversion) were significantly better in patients who additionally took delamanid.[3][4]
The European Medicines Agency (EMA) recommended conditional marketing authorization for delamanid in adults with multidrug-resistant pulmonary tuberculosis without other treatment options because of resistance or tolerability. The EMA considered the data show that the benefits of delamanid outweigh the risks, but that additional studies were needed on the long-term effectiveness.[5]
Delamanid, an antibiotic active against Mycobacterium tuberculosis strains, has been filed for approval in the E.U. and by Otsuka for the treatment of multidrug-resistant tuberculosis. In 2013, a positive opinion was received in the E.U. for this indication. Phase III trials for treatment of multidrug-resistant tuberculosis are under way in the U.S. Phase II study for the pediatric use is undergone in the Europe.
The drug candidate’s antimycobacterial mechanism of action is via specific inhibition of the synthesis pathway of mycolic acid, which is a cell wall component unique to M. tuberculosis.
In 2008, orphan drug designation was received in Japan for the treatment of pulmonary tuberculosis.
![]()
Tuberculosis (TB), an airborne lung infection, still remains a major public health problem worldwide. It is estimated that about 32% of the world population is infected with TB bacillus, and of those, approximately 8.9 million people develop active TB and 1.7 million die as a result annually according to 2004 figures. Human immunodeficiency virus (HIV) infection has been a major contributing factor in the current resurgence of TB. HIV-associated TB is widespread, especially in sub-Saharan Africa, and such an infectious process may further accelerate the resurgence of TB.
Moreover, the recent emergence of multidrug-resistant (MDR) strains ofMycobacterium tuberculosis that are resistant to two major effective drugs, isonicotinic acid hydrazide (INH) and rifampicin (RFP), has further complicated the world situation.
The World Health Organization (WHO) has estimated that if the present conditions remain unchanged, more than 30 million lives will be claimed by TB between 2000 and 2020. As for subsequent drug development, not a single new effective compound has been launched as an antituberculosis agent since the introduction of RFP in 1965, despite the great advances that have been made in drug development technologies.
Although many effective vaccine candidates have been developed, more potent vaccines will not become immediately available. The current therapy consists of an intensive phase with four drugs, INH, RFP, pyrazinamide (PZA), and streptomycin (SM) or ethambutol (EB), administered for 2 months followed by a continuous phase with INH and RFP for 4 months. Thus, there exists an urgent need for the development of potent new antituberculosis agents with low-toxicity profiles that are effective against both drug-susceptible and drug-resistant strains of M. tuberculosis and that are capable of shortening the current duration of therapy.
………………………
US20060094767
(R)-2-bromo-4-nitro-1-(2-methyl-2-oxiranylmethyl)imidazole
4-[4-(4-Trifluoromethoxyphenoxy)piperidin-1-yl]phenol
ARE THE INTERMEDIATES
Example 1884
Production of (R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole
4-[4-(4-Trifluoromethoxyphenoxy)piperidin-1-yl]phenol (693 mg, 1.96 mmol) was dissolved in N,N′-dimethylformamide (3 ml), and sodium hydride (86 mg, 2.16 mmol) was added while cooling on ice followed by stirring at 70-75° C. for 20 minutes. The mixture was cooled on ice. To the solution, a solution prepared by dissolving (R)-2-bromo-4-nitro-1-(2-methyl-2-oxiranylmethyl)imidazole (720 mg, 2.75 mmol) in N,N′-dimethylformamide (3 ml) was added followed by stirring at 70-75° C. for 20 minutes. The reaction mixture was allowed to return to room temperature, ice water (25 ml) was added, and the resultant solution was extracted with methylene chloride (50 ml) three times. The organic phases were combined, washed with water 3 times, and dried over magnesium sulfate. After filtration, the filtrate was concentrated, and the residue was purified by silica gel column chromatography (methylene chloride/ethyl acetate=3/1). Recrystallization from ethyl acetate/isopropyl ether gave (R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole (343 mg, 33%) as a light yellow powder.
…………………………
WO 2010021409 AND http://worldwide.espacenet.com/publicationDetails/biblio?CC=IN&NR=203704A1&KC=A1&FT=D
FOR 2, 4 DINITROIMIDAZOLE
…………………………………………
WO2011093529A1
These patent literatures disclose Reaction Schemes A and B below as the processes for producing the aforementioned 2, 3-dihydroimidazo [2, 1-b] oxazole compound.
Reaction Scheme A:
wherein R1 is a hydrogen atom or lower-alkyl group; R2 is a substituted pxperidyl group or a substituted piperazinyl group; and X1 is a halogen atom or a nitro group.
Reaction Scheme B:
wherein X2 is a halogen or a group causing a substitution reaction similar to that of a halogen; n is an integer from 1 to 6; and R1, R2 and X1 are the same as in Reaction Scheme A.
An oxazole com ound represented by Formula (la) :
, i.e., 2-methyl-6-nitro-2-{4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl }-2, 3- dihydroimidazo [2, 1-b] oxazole (hereunder, this compound may be simply referred to as “Compound la”) is produced, for example, by the method shown in the Reaction Scheme C below (Patent
Literature 3) . In this specification, the term “oxazole compound’ means an oxazole derivative that encompasses compounds that contain an oxazole ring or an oxazoline ring (dihydrooxazole ring) in the molecule.
Reaction Scheme C:
However, the aforementioned methods are unsatisfactory in terms of the yield of the objective compound. For example, the method of Reaction Scheme C allows the objective oxazole Compound (la) to be obtained from Compound (2a) at a yield as low as 35.9%. Therefore, alternative methods for producing the compound in an industrially advantageous manner are desired. Citation List
Patent Literature
PTL 1: WO2004/033463
PTL 2: WO2004/035547
PTL 3: WO2008/140090
Example 9
Production of (R) -2-methyl-6-nitro-2- { 4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl } -2 , 3- dihydroimidazo [2 , 1-b] oxazole
{R) -1- [ - {2 , 3-epoxy-2-methylpropoxy ) phenyl] -4- [4- ( trifluoromethoxy ) phenoxy ] piperidine (10.0 g, 23.6 mmol, optical purity of 94.3%ee), 2-chloro-4-nitroimidazole (4.0 g, 27.2 mmol), sodium acetate (0.4 g, 4.9 mmol), and t- butyl acetate (10 ml) were mixed and stirred at 100°C for 3.5 hours. Methanol (70 ml) was added to the reaction mixture, and then a 25% sodium hydroxide aqueous solution (6.3 g, 39.4 mmol) was added thereto dropwise while cooling with ice. The resulting mixture was stirred at 0°C for 1.5 hours, and further stirred at approximately room
temperature for 40 minutes. Water (15 ml) and ethyl acetate (5 ml) were added thereto, and the mixture was stirred at 45 to 55°C for 1 hour. The mixture was cooled to room temperature, and the precipitated crystals were collected by filtration. The precipitated crystals were subsequently washed with methanol (30 ml) and water (40 ml) . Methanol (100 ml) was added to the resulting
crystals, followed by stirring under reflux for 30 minutes. The mixture was cooled to room temperature. The crystals were then collected by filtration and washed with methanol (30 ml) . The resulting crystals were dried under reduced pressure, obtaining 9.3 g of the objective product (yield: 73%) .
Optical purity: 99.4%ee.
……………….
Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles
J Med Chem 2006, 49(26): 7854
http://pubs.acs.org/doi/abs/10.1021/jm060957y
(R)-2-Methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole (19, DELAMANID).
To a mixture of 27 (127.56 g, 586.56 mmol) and 4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenol (28g) (165.70 g, 468.95 mmol) in N,N-dimethylformamide (1600 mL) was added 60% sodium hydride (22.51 g, 562.74 mmol) at 0 °C portionwise. After the mixture was stirred at 50 °C for 2 h under a nitrogen atmosphere, the reaction mixture was cooled in an ice bath and carefully quenched with ethyl acetate (230 mL) and ice water (50 mL). The thus-obtained mixture was poured into water (3000 mL) and stirred for 30 min. The resulting precipitates were collected by filtration, washed with water, and dried at 60 °C overnight. This crude product was purified by silica gel column chromatography using a dichloromethane and ethyl acetate mixture (5/1) as solvent. The appropriate fractions were combined and evaporated under reduced pressure. The residue was recrystallized from ethyl acetate (1300 mL)−isopropyl alcohol (150 mL) to afford 19 (119.11 g, 48%) as a pale yellow crystalline powder.
Mp 195−196 °C.
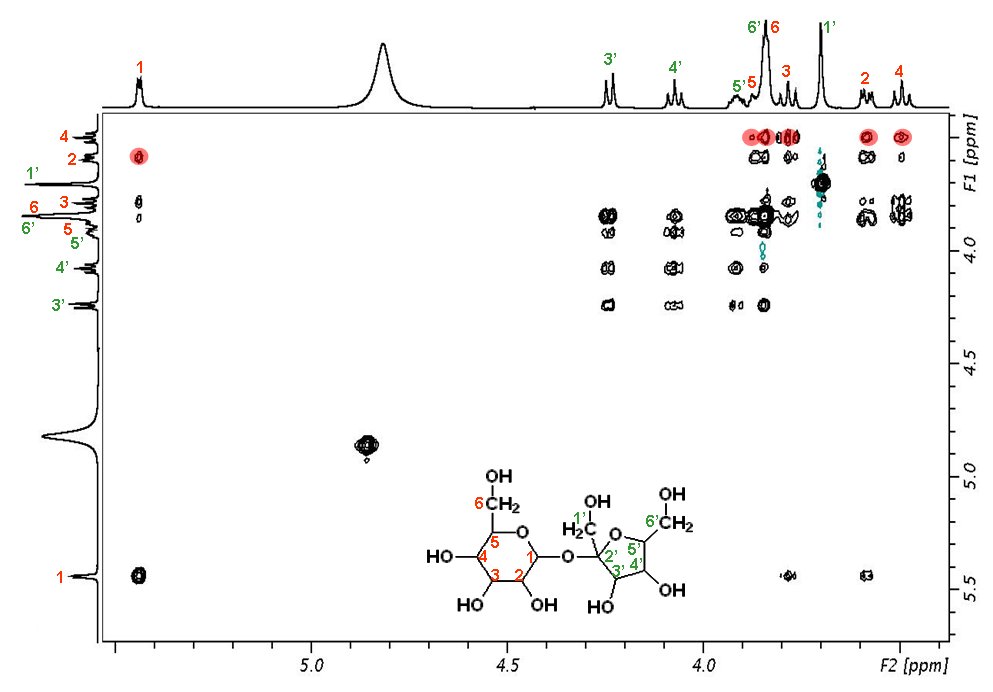
1H NMR (CDCl3) δ 1.77 (3H, s), 1.87−2.16 (4H, m), 2.95−3.05 (2H, m), 3.32−3.41 (2H, m), 4.02 (1H, d, J = 10.2 Hz), 4.04 (1H, d, J = 10.2 Hz), 4.18 (1H, J = 10.2 Hz), 4.36−4.45 (1H, m), 4.49 (1H, d, J = 10.2 Hz), 6.76 (2H, d, J = 6.7 Hz), 6.87−6.94 (4H, m), 7.14 (2H, d, J = 8.6 Hz), 7.55 (1H, s).
[α ![]() −9.9° (c 1.01, CHCl3).
−9.9° (c 1.01, CHCl3).
MS (DI) m/z 535 (M+ + 1). Anal. (C25H25F3N4O6) C, H, N.
http://pubs.acs.org/doi/suppl/10.1021/jm060957y/suppl_file/jm060957ysi20061113_095044.pdf
References
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. (2006). "OPC-67683, a Nitro-Dihydro-Imidazooxazole Derivative with Promising Action against Tuberculosis in Vitro and in Mice". PLoS Medicine 3 (11): e466.doi:10.1371/journal.pmed.0030466. PMC 1664607. PMID 17132069. edit
- Skripconoka, V.; Danilovits, M.; Pehme, L.; Tomson, T.; Skenders, G.; Kummik, T.; Cirule, A.; Leimane, V.; Kurve, A.; Levina, K.; Geiter, L. J.; Manissero, D.; Wells, C. D. (2012). "Delamanid Improves Outcomes and Reduces Mortality for Multidrug-Resistant Tuberculosis". European Respiratory Journal41 (6): 1393–1400. doi:10.1183/09031936.00125812. PMC 3669462. PMID 23018916. edit
- H. Spreitzer (18 February 2013). "Neue Wirkstoffe – Bedaquilin und Delamanid". Österreichische Apothekerzeitung (in German) (4/2013): 22.
- Gler, M. T.; Skripconoka, V.; Sanchez-Garavito, E.; Xiao, H.; Cabrera-Rivero, J. L.; Vargas-Vasquez, D. E.; Gao, M.; Awad, M.; Park, S. K.; Shim, T. S.; Suh, G. Y.; Danilovits, M.; Ogata, H.; Kurve, A.; Chang, J.; Suzuki, K.; Tupasi, T.; Koh, W. J.; Seaworth, B.; Geiter, L. J.; Wells, C. D. (2012). "Delamanid for Multidrug-Resistant Pulmonary Tuberculosis". New England Journal of Medicine 366 (23): 2151–2160. doi:10.1056/NEJMoa1112433.PMID 22670901. edit
- Drug Discovery & Development. EMA Recommends Two New Tuberculosis Treatments. November 22, 2013.
- Synthesis and antituberculous activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles
45th Intersci Conf Antimicrob Agents Chemother (ICAAC) (December 16-19, Washington DC) 2005, Abst F-1473
|
|
12-28-2006
|
Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles.
|
Journal of medicinal chemistry
|
|
|
11-1-2006
|
OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice.
|
PLoS medicine
|
|
|
1-1-2008
|
New anti-tuberculosis drugs with novel mechanisms of action.
|
Current medicinal chemistry
|
|
|
11-11-2010
|
Synthesis and Structure-Activity Relationships of Aza- and Diazabiphenyl Analogues of the Antitubercular Drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824).
|
Journal of medicinal chemistry
|
|
|
5-1-2012
|
Tuberculosis: the drug development pipeline at a glance.
|
European journal of medicinal chemistry
|
|
|
1-12-2012
|
Structure-activity relationships for amide-, carbamate-, and urea-linked analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824).
|
Journal of medicinal chemistry
|
|
|
9-11-2009
|
Pharmaceutical Composition Achieving Excellent Absorbency of Pharmacologically Active Substance
|
|
|
1-16-2009
|
Sulfonamide Derivatives for the Treatment of Bacterial Infections
|
| WO2004033463A1 |
Oct 10, 2003 |
Apr 22, 2004 |
Otsuka Pharma Co Ltd |
2,3-DIHYDRO-6-NITROIMIDAZO[2,1-b]OXAZOLES |
| WO2004035547A1 |
Oct 14, 2003 |
Apr 29, 2004 |
Otsuka Pharma Co Ltd |
1-substituted 4-nitroimidazole compound and process for producing the same |
| WO2008140090A1 |
May 7, 2008 |
Nov 20, 2008 |
Otsuka Pharma Co Ltd |
Epoxy compound and method for manufacturing the same |
| JP2009269859A * |
|
|
|
Title not available |
TB
![Figure]()
It is estimated that a third of the world’s population is currently infected with tuberculosis, leading to 1.6 million deaths annually. The current drug regimen is 40 years old and takes 6-9 months to administer. In addition, the emergence of drug resistant strains and HIV co-infection mean that there is an urgent need for new anti-tuberculosis drugs. The twenty-first century has seen a revival in research and development activity in this area, with several new drug candidates entering clinical trials. This review considers new potential first-line anti-tuberculosis drug candidates, in particular those with novel mechanisms of action, as these are most likely to prove effective against resistant strains.
From among acid-fast bacteria, human Mycobacterium tuberculosis has been widely known. It is said that the one-third of the human population is infected with this bacterium. In addition to the human Mycobacterium tuberculosis, Mycobacterium africanum and Mycobacterium bovis have also been known to belong to the Mycobacterium tuberoculosis group. These bacteria are known as Mycobacteria having a strong pathogenicity to humans.
Against these tuberculoses, treatment is carried out using three agents, rifampicin, isoniazid, and ethambutol (or streptomycin) that are regarded as first-line agents, or using four agents such as the above three agents and pyrazinamide.
However, since the treatment of tuberculosis requires extremely long-term administration of agents, it might result in poor compliance, and the treatment often ends in failure.
Moreover, in respect of the above agents, it has been reported that: rifampicin causes hepatopathy, flu syndrome, drug allergy, and its concomitant administration with other drugs is contraindicated due to P450-associated enzyme induction; that isoniazid causes peripheral nervous system disorder and induces serious hepatopathy when used in combination with rifampicin; that ethambutol brings on failure of eyesight due to optic nerve disorder; that streptomycin brings on diminution of the hearing faculty due to the 8th cranial nerve disorder; and that pyrazinamide causes adverse reactions such a hepatopathy, gouty attack associated with increase of uric acid level, vomiting (A Clinician’s Guide To Tuberculosis, Michael D. Iseman 2000 by Lippincott Williams & Wilkins, printed in the USA, ISBN 0-7817-1749-3, Tuberculosis, 2nd edition, Fumiyuki Kuze and Takahide Izumi, Igaku-Shoin Ltd., 1992).
Actually, it has been reported that cases where the standard chemotherapy could not be carried out due to the adverse reactions to these agents made up 70% (approximately 23%, 52 cases) of the total cases where administration of the agents was discontinued (the total 228 hospitalized patients who were subject to the research) (Kekkaku, Vol. 74, 77-82, 1999).
In particular, hepatotoxicity, which is induced by rifampicin, isoniazid, and ethambutol out of the 5 agents used in combination for the aforementioned first-line treatment, is known as an adverse reaction that is developed most frequently. At the same time, Mycobacterium tuberculosis resistant to antitubercular agents, multi-drug-resistant Mycobacterium tuberculosis, and the like have been increasing, and the presence of these types of Mycobacterium tuberculosismakes the treatment more difficult.
According to the investigation made by WHO (1996 to 1999), the proportion ofMycobacterium tuberculosis that is resistant to any of the existing antitubercular agents to the total types of Mycobacterium tuberculosis that have been isolated over the world reaches 19%, and it has been published that the proportion of multi-drug-resistant Mycobacterium tuberculosis is 5.1%. The number of carriers infected with such multi-drug-resistant Mycobacterium tuberculosis is estimated to be 60,000,000, and concerns are still rising that multi-drug-resistantMycobacterium tuberculosis will increase in the future (April 2001 as a supplement to the journal Tuberculosis, the “Scientific Blueprint for TB Drug Development.”)
In addition, the major cause of death of AIDS patients is tuberculosis. It has been reported that the number of humans suffering from both tuberculosis and HIV reaches 10,700,000 at the time of year 1997 (Global Alliance for TB drug development). Moreover, it is considered that the mixed infection of tuberculosisand HIV has an at least 30 times higher risk of developing tuberculosis than the ordinary circumstances.
Taking into consideration the aforementioned current situation, the profiles of the desired antitubercular agent is as follows: (1) an agent, which is effective even for multi-drug-resistant Mycobacterium tuberculosis, (2) an agent enabling a short-term chemotherapy, (3) an agent with fewer adverse reactions, (4) an agent showing an efficacy to latent infecting Mycobacterium tuberculosis (i.e., latentMycobacterium tuberculosis), and (5) an orally administrable agent.
Examples of bacteria known to have a pathogenicity to humans include offending bacteria of recently increasing MAC infection (Mycobacterium avium—intracellulare complex infection) such as Mycobacterium avium andMycobacterium intracellulare, and atypical acid-fast bacteria such asMycobacterium kansasii, Mycobacterium marinum, Mycobacterium simiae, Mycobacterium scrofulaceum, Mycobacterium szulgai, Mycobacterium xenopi, Mycobacterium malmoense, Mycobacterium haemophilum, Mycobacterium ulcerans, Mycobacterium shimoidei, Mycobacterium fortuitum, Mycobacterium chelonae, Mycobacterium smegmatis, and Mycobacterium aurum.
Nowadays, there are few therapeutic agents effective for these atypical acid-fast bacterial infections. Under the presence circumstances, antitubercular agents such as rifampicin, isoniazid, ethambutol, streptomycin and kanamycin, a newquinolone agent that is a therapeutic agent for common bacterial infections, macrolide antibiotics, aminoglycoside antibiotics, and tetracycline antibiotics are used in combination.
However, when compared with the treatment of common bacterial infections, the treatment of atypical acid-fast bacterial infections requires a long-term administration-of agents, and there have been reported cases where the infection is changed to an intractable one, finally leading to death. To break the afore-mentioned current situation, the development of an agent having a stronger efficacy is desired.
For example, National Publication of International Patent Application No. 11-508270 (WO97/01562) discloses that a 6-nitro-1,2,3,4-tetrahydro[2,1-b]-imidazopyran compound has a bactericidal action in vitro to Mycobacterium tuberculosis (H37Rv strain) and multi-drug-resistant Mycobacterium tuberculosis, and that the above compound has a therapeutic effect to a tuberculosis-infected animal model when it is orally administered and thus useful as antitubercular agent.
![Share]()













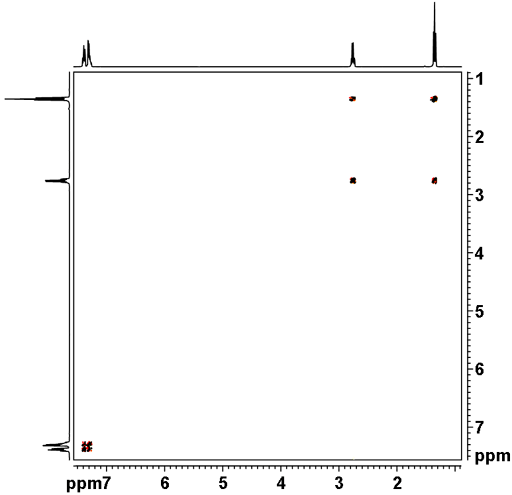

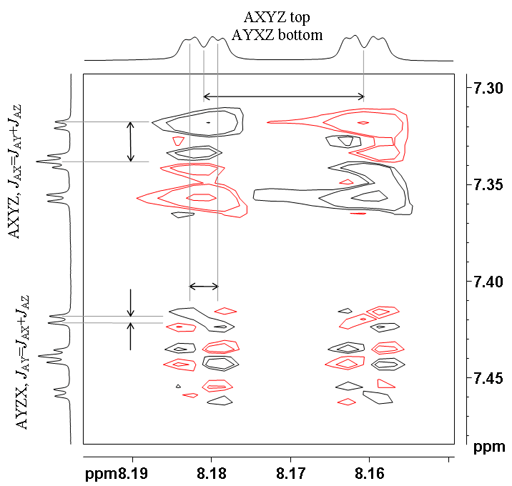
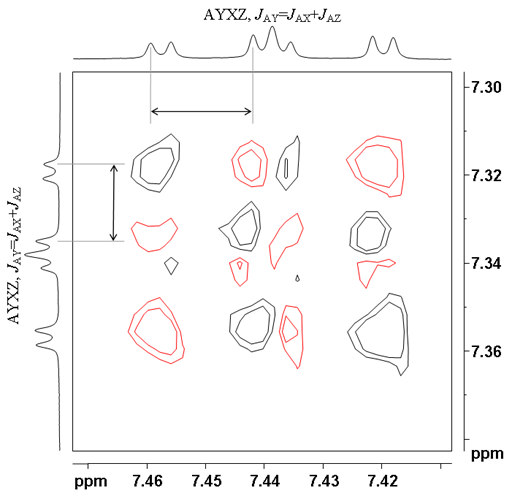
![12,14-ditbutylbenzo[g]chrysene](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/dtbbgc.gif)
![COSY of 12,14-ditbutylbenzo[g]chrysene](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/cosydtbbgc.gif)
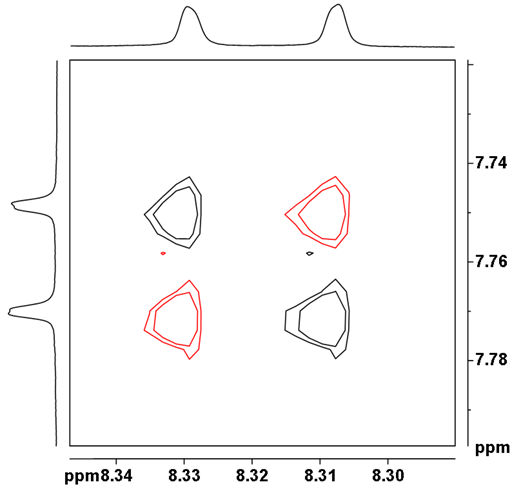
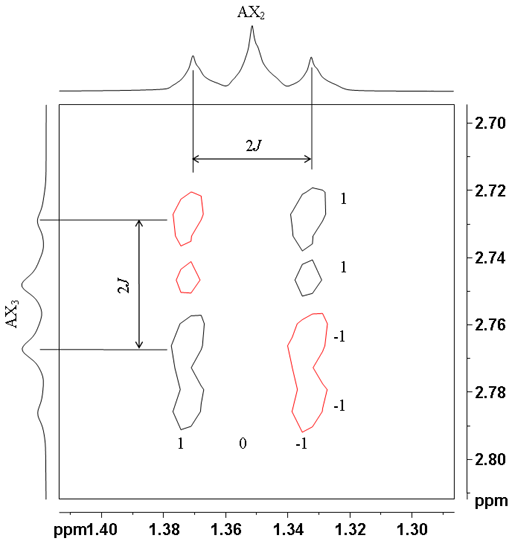
![Aromatic region of COSY of 12,14-ditbutylbenzo[g]chrysene](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/aromaticdtbbgc.gif)
![Aromatic region of COSY of 12,14-ditbutylbenzo[g]chrysene](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/aromaticdtbbgc2.gif)
![Aromatic region of COSY of 12,14-ditbutylbenzo[g]chrysene in color](http://chem.ch.huji.ac.il/nmr/techniques/2d/cosy/cosy_files/coloraromatic.gif)







 Relugolix (TAK-385)
Relugolix (TAK-385)


 tak 385
tak 385


 The experimental drug was developed by Prof Frank Longo from Stanford University
The experimental drug was developed by Prof Frank Longo from Stanford University














































![Graphical abstract: Radiolabelling of 1,4-disubstituted 3-[18F]fluoropiperidines and its application to new radiotracers for NR2B NMDA receptor visualization](http://pubs.rsc.org/services/images/RSCpubs.ePlatform.Service.FreeContent.ImageService.svc/ImageService/image/GA?id=C2OB26378E "Graphical abstract")


 is formed by polymerization of doxorubicin- and camptothecin-derivatized monomers and a cisplatin cross-linker.")














 −9.9° (c 1.01, CHCl3).
−9.9° (c 1.01, CHCl3).